This web page was produced as an assignment for Gen677 at UW-Madison Spring 2013
Protein Phylogeny
A phylogeny is the evolutionary history of an entire group of organisms. A phylogenetic tree is a visual representation of this evolutionary history. A phylogeny can be determined based on morphological or molecular traits and tracks how a group of species evolves. The goal of phylogenetic analysis is to determine the branching of different lineages over evolutionary time. The following analysis focuses on protein phylogeny which is based on the comparison of protein sequences to determine their relatedness.
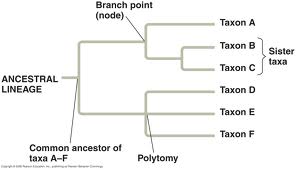
Figure 1. Example of a Phylogenetic Tree. Phylogenetic trees show the lineage of taxa and how they relate to each other. The root is the common ancestor of all taxa shown. Branch points show the point of last common ancestry between different taxa.
The Clustal Omega trees were made using two different methods, neighbor joining and average distance. The neighbor joining method looks at the distance between each sequence compared, groups together the most closely related sequences and tries to minimize total branch length [1]. The average distance method uses sequence alignments to determine distances and create a tree based off of those distances [2].
MYO5a Protein Phylogeny
In order to create phylogenetic trees the human MYO5a protein was compared with nine vertebrate and five invertebrate species. The alignments used to create the trees were made using Clustal Omega and T-Coffee [3,4,5].
Clustal Omega Trees
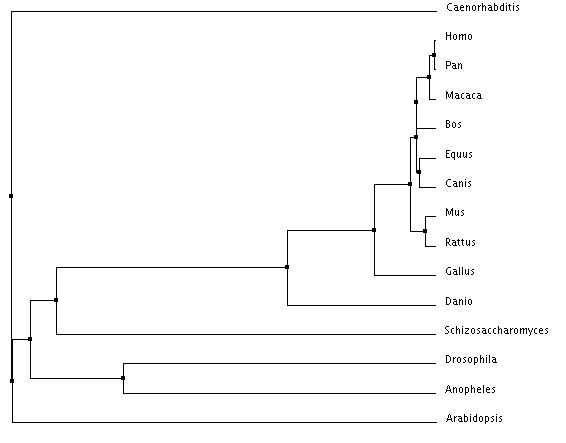
Figure 2. Average Distance Tree Using PID
|
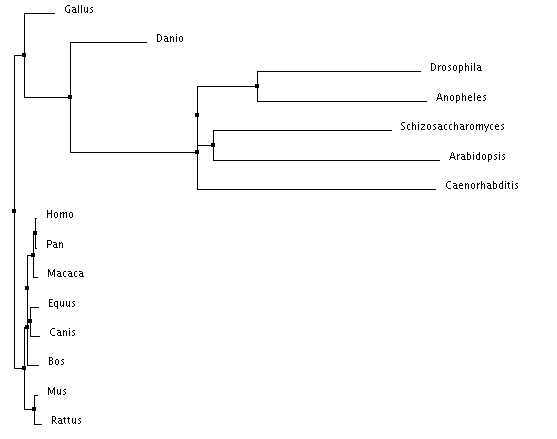
Figure 3. Neighbor Joining Tree Using PID
|
Phylogeny.fr Tree via T-Coffee [1][2][3]
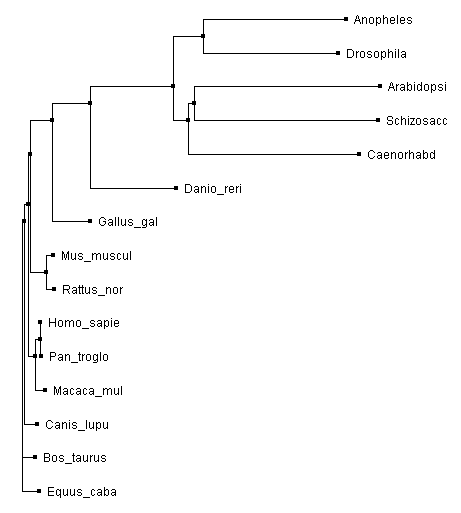
Figure 4. Rooted Phylogram
Analysis and Discussion
The trees made using Clustal Omega and T-Coffee both show the invertebrates acting as an out group for the highly conserved vertebrate MYO5a. The neighbor joining tree and the rooted phylogram are in complete agreement on the placement of the species in relation to each other. The average distance tree is the only tree with a placement disagreement. The average distance tree places yeast closer to the vertebrates then to the invertebrates while the other two trees place yeast in close association with arabidopsis.
References
[1] Saitou N., Nei M. (1987) The neighbor-joining method: a method for reconstructing phylogenetic trees. Mol Biol Evol. 4(4):406-25.
[2] Mount , D. M. (2004) Bioinformatics: Sequence and Genome Analysis 2nd ed. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY
[3] Dereeper A., Audic S., Claverie J.M., Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010 Jan 12;10:8. (PubMed)
[4] Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M., Claverie J.-M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist Nucleic Acids Research. 2008 Jul 1; 36 (Web Server Issue):W465-9. Epub 2008 Apr 19. (PubMed)
[5] Notredame C., Higgins DG., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000, Sep 8;302(1):205-17. (PubMed)
[2] Mount , D. M. (2004) Bioinformatics: Sequence and Genome Analysis 2nd ed. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY
[3] Dereeper A., Audic S., Claverie J.M., Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010 Jan 12;10:8. (PubMed)
[4] Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M., Claverie J.-M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist Nucleic Acids Research. 2008 Jul 1; 36 (Web Server Issue):W465-9. Epub 2008 Apr 19. (PubMed)
[5] Notredame C., Higgins DG., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000, Sep 8;302(1):205-17. (PubMed)
